Physics predicts why and where biological cells are regulated
It can seem that cells make choices about behavior that is often hard to explain, but often mimic social interactions. Game theory, in particular, has been insightful for understanding the dynamics of populations of microbes. Unfortunately, an unintended consequence of applying social concepts such as game theory to cells is the use of language that frames our thinking. Bet hedging strategies, cooperators, cheaters, noncooperators, altruistic cells, etc. are common descriptions of cell behavior found in the literature. Who doesn’t think of a cancer cell as being a selfish cell – undergoing unlimited growth despite the fact that it will ultimately kill its host? But this anthropomorphic view doesn’t help us cure cancer, either. (Image courtesy of Nathan Johnson and Pacific Northwest National Laboratory).
In comparison, physics-based theories about life, which have been around for 100 years, have garnered little attention. Lotka proposed in 1922 that natural selection is about who can harvest the required energy for reproduction from the environment the fastest. We now know that biological cells are part of the same phenomena as hurricanes and tornadoes, called dissipative systems. As the lower atmosphere heats up in the summer due to radiation from the sun, winds form that move the hot air to the cooler upper atmosphere. Under normal conditions, the winds are relatively random – a gust here, a gust there and seemingly changing directions from moment to moment. But when the temperature difference between the upper and lower atmosphere becomes large enough, the winds become correlated in an attempt to reduce the temperature difference as quick as possible. Circular wind patterns form, with heat being taken from the hot lower atmosphere and dumped in the cooler upper atmosphere. When these circular winds rotate sideways, they become tornadoes.
From the viewpoint of physics, the atmosphere is trying to redistribute energy equally. The fastest way to do that is to have correlated motions (convection cells) to do that.
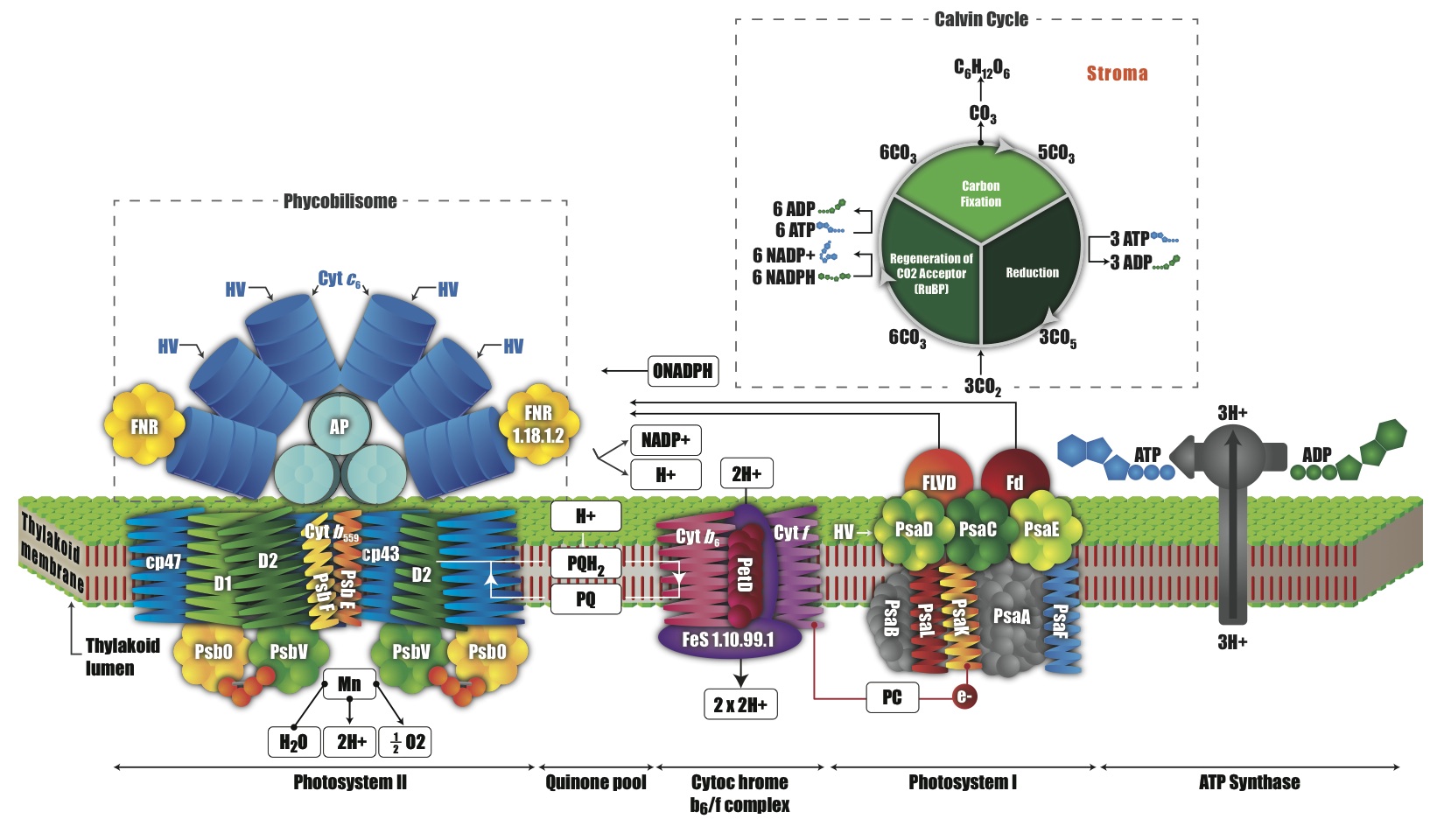
Biological cells serve the same purpose, but now the correlated processes are enabled by visible chemical structures. Sunlight comes to the cell in the form of high energy radiation, and that radiation is captured and turned into lower energy chemical compounds. The driving force again is the need to distribute energy equally in nature. Except now, those lower energy chemical compounds form the cell structures needed to capture the sunlight. The more sun that is captured by a cell, the more cells that can be produced – to capture more sunlight. This is the process of metabolism and growth.
The chemical compounds produced need to be in just the right proportions to form structures that can capture energy, however. This is where things can get out of control. If some compounds are produced in too great of a quantity, the energy-capturing structures can’t form. Cells regulate this process. Dysregulation can cause more than just cells not working well – it can cause cancer – unregulated growth to the detriment of the host. In our new study, we have shown that known sites of metabolic regulation in cells, enzyme regulation, can be predicted based on these very principles.
Read about the technical details in the article, Enzyme activities predicted by metabolite concentrations and solvent capacity in the cell, published in the Journal of The Royal Society Interface https://royalsocietypublishing.org/doi/10.1098/rsif.2020.0656. This work was paid for by the National Institute of Biomedical Imaging and Bioengineering and the U.S. Department of Energy, Office of Biological and Environmental Research.